Figure 2-31
Electron micrograph of fibroblast cytoplasm. Note the actin filaments (AF) and microtubules (MT). x60,000. (Courtesy of E Katchburian.)
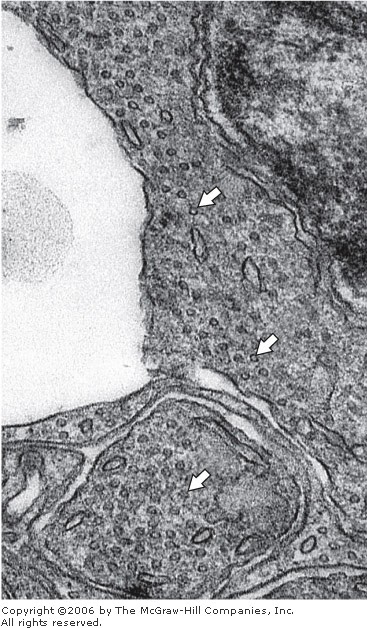
Figure 2-32
Electron micrograph of a section of a photosensitive retinal cell. Note the accumulation of transversely sectioned microtubules (arrows). Reduced slightly from x80,000.
Figure 2-33
Photomicrograph of the epithelium covering the inner surface of the respiratory airways. Most cells in this epithelium contain numerous cilia in their apices (free upper extremities). N, cell nuclei; M, cytoplasmic mucus secretion, which appears dark in this preparation. H&E stain. High magnification.
Figure 2-34

Schematic representation of microtubules, cilia, and centrioles. A: Microtubules as seen in the electron microscope after fixation with tannic acid in glutaraldehyde. The unstained tubulin subunits are delineated by the dense tannic acid. Cross sections of tubules reveal a ring of 13 subunits of dimers arranged in a spiral. Changes in microtubule length are due to the addition or loss of individual tubulin subunits. B: A cross section through a cilium reveals a core of microtubules called an axoneme. The axoneme consists of two central microtubules surrounded by nine microtubule doublets. In the doublets, microtubule A is complete and consists of 13 subunits, whereas microtubule B shares two or three heterodimers with A. When activated by ATP, the dynein arms link adjacent tubules and provide for the sliding of doublets against each other. C: Centrioles consist of nine microtubule triplets linked together in a pinwheel-like arrangement. In the triplets, microtubule A is complete and consists of 13 subunits, whereas microtubules B and C share tubulin subunits. Under normal circumstances, these organelles are found in pairs with the centrioles disposed at right angles to one another.
The subunit of a microtubule is a heterodimer composed of

and
 tubulin
tubulin molecules of closely related amino acid composition, each with a molecular mass of about 50 kDa.
Under appropriate conditions (in vivo or in vitro), tubulin subunits polymerize to form microtubules. With special staining procedures, tubulin can be seen as heterodimers organized into a spiral. A total of 13 units is present in one complete turn of the spiral (Figure 2–34).
Polymerization of tubulins to form microtubules in vivo is directed by a variety of structures collectively known as microtubule-organizing centers. These structures include cilia, basal bodies, and centrosomes. Microtubule growth, via subunit polymerization, occurs more rapidly at one end of existing microtubules. This end is referred to as the plus (+) end, and the other extremity is the minus (–) end. Tubulin polymerization is under control of the concentration of Ca2+ and of the microtubule-associated proteins, or MAPs. Microtubule stability is variable; for example, microtubules of cilia are stable, whereas microtubules of the mitotic spindle have a short duration. The antimitotic alkaloid colchicine binds specifically to tubulin, and when the complex tubulin–colchicine binds to microtubules, it prevents the addition of more tubulin in the plus (+) extremity. Mitotic microtubules are broken down because the depolymerization continues, mainly at the minus (–) end, and the lost tubulin units are not replaced. Another alkaloid that interferes with the mitotic microtubule is taxol, which accelerates the formation of microtubules but at the same time stabilizes them. All cytosolic tubulin is used in stable microtubules, and no tubulin is left for the formation of the mitotic spindle. Another alkaloid, vinblastine, acts by depolymerizing formed microtubules and, in a second step, aggregating to form paracrystalline arrays of tubulin.
Medical Application
The antimitotic alkaloids are useful tools in cell biology (eg, colchicine is used to arrest chromosomes in metaphase and to prepare karyotypes) and in cancer chemotherapy (eg, vinblastine, vincristine, and taxol are used to arrest cell proliferation in tumors). Because tumor cells proliferate rapidly, they are more affected by antimitotic drugs than are normal cells. However, chemotherapy has many undesirable consequences. For example, some normal blood-forming cells and the epithelial cells that cover the digestive tract also show a high rate of proliferation and are adversely affected by chemotherapy.
Cytoplasmic microtubules are stiff structures that play a significant role in the development and maintenance of cell shape. They are usually present in a proper orientation, either to effect development of a given cellular asymmetry or to maintain it. Procedures that disrupt microtubules result in the loss of this cellular asymmetry.
Microtubules also participate in the intracellular transport of organelles and vesicles. Examples include axoplasmic transport in neurons, melanin transport in pigment cells, chromosome movements by the mitotic spindle, and vesicle movements among different cell compartments. In each of these examples, movement is related to the presence of complex microtubule networks, and such activities are suspended if microtubules are disrupted. The transport guided by microtubules is under the control of special proteins called motor proteins, which use energy to move molecules and vesicles.
Microtubules provide the basis for several complex cytoplasmic components, including centrioles, basal bodies, cilia, and flagella.
Centrioles are cylindrical structures (0.15

m in diameter and 0.3–0.5
m in length) composed primarily of short, highly organized microtubules (Figure 2–34). Each centriole shows nine sets of microtubules arranged in triplets. The microtubules are so close together that adjacent microtubules of a triplet share a common wall. Close to the nucleus of nondividing cells is a
centrosome (Figure 2–35) made of a pair of centrioles surrounded by a granular material. In each pair, the long axes of the centrioles are at right angles to each other. Before cell division, more specifically during the S period of the interphase, each centrosome duplicates itself so that now each centrosome has two pairs of centrioles. During mitosis, the centrosomes divide in two, move to opposite poles of the cell, and become organizing centers for the microtubules of the mitotic spindle.
Figure 2-35
Drawing of a centrosome with its granular protein material surrounding a pair of centrioles, one shown at a right angle to the other. Each centriole is composed of nine bundles of microtubules, with three microtubules per bundle.
Cilia and
flagella (singular, cilium, flagellum) are motile processes, covered by cell membrane, with a highly organized microtubule core. Ciliated cells typically possess a large number of cilia, each about 2–3
m in length. Flagellated cells have only one flagellum, with a length close to 100
m. In humans, the spermatozoa are the only cell type with a flagellum. The main function of cilia is to sweep fluid from the surface of cell sheets. Both cilia and flagella possess the same core organization.
This core consists of nine pairs of microtubules surrounding two central microtubules. This sheaf of microtubules, possessing a 9 + 2 pattern, is called an axoneme (Gr. axon, axis, + nema, thread). Each of the nine peripheral pairs shares a common wall (Figure 2–34). The microtubules in the central pair are enclosed within a central sheath. Adjacent peripheral pairs are linked to each other by protein bridges called nexins and to the central sheath by radial spokes. The microtubules of each pair are identified as A and B. Microtubule A is complete, with 13 heterodimers, whereas B has only 10 heterodimers (in a cross section). Extending from the surface of microtubule A are pairs of arms formed by the protein dynein, which has ATPase activity.
At the base of each cilium or flagellum is a basal body, essentially similar to a centriole, that controls the assembly of the axoneme.
Medical Application
Several mutations have been described in the proteins of the cilia and flagella. They are responsible for the immotile cilia syndrome, the symptoms of which are immotile spermatozoa, male infertility, and chronic respiratory infections caused by the lack of the cleansing action of cilia in the respiratory tract.
References
| Afzelius BA, Eliasson R: Flagellar mutants in man: on the heterogeneity of the immotile-cilia syndrome. J Ultrastruct Res 1979;69:43. [PMID: 501788] |
| Aridor M, Balch WE: Integration of endoplasmic reticulum signaling in health and disease. Nat Med 1999;5:745. [PMID: 10395318] |
| Barrit GJ: Communication Within Animal Cells. Oxford University Press, 1992. |
| Becker WM et al: The World of the Cell, 4th ed. Benjamin/Cummings, 2000. |
| Bretscher MS: The molecules of the cell membrane. Sci Am 1985;253:100. [PMID: 2416050] |
| Brinkley BR: Microtubule organizing centers. Annu Rev Cell Biol 1985;1:145. [PMID: 3916316] |
| Brown MS et al: Recycling receptors: the round-trip itinerary of migrant membrane proteins. Cell 1983;32:663. [PMID: 6299572] |
| Cooper GM: The Cell: A Molecular Approach. ASM Press/Sinauer Associates, Inc., 1997. |
| DeDuve C: A Guided Tour of the Living Cell. Freeman, 1984. |
| DeDuve C: Microbodies in the living cell. Sci Am 1983;248:74. |
| Dustin P: Microtubules, 2nd ed. Springer-Verlag, 1984. |
| Farquhar MG: Progress in unraveling pathways of Golgi traffic. Annu Rev Cell Biol 1985;1:447. [PMID: 3916320] |
| Fawcett D: The Cell, 2nd ed. Saunders, 1981. |
| Krstíc RV: Ultrastructure of the Mammalian Cell. Springer-Verlag, 1979. |
| Mitchison TJ, Cramer LP: Actin-based cell motility and cell locomotion. Cell 1996;84:371. [PMID: 8608590] |
| Osborn M, Weber K: Intermediate filaments: cell-type-specific markers in differentiation and pathology. Cell 1982;31:303. [PMID: 6891619] |
| Pfeffer SR, Rothman JE: Biosynthetic protein transport and sorting in the endoplasmic reticulum. Annu Rev Biochem 1987;56:829. [PMID: 3304148] |
| Rothman J: The compartmental organization of the Golgi apparatus. Sci Am 1985;253:74. [PMID: 3929377] |
| Simons K, Ikonen E: How cells handle cholesterol. Science 2000;290:1721. [PMID: 11099405] |
| Tzagoloff A: Mitochondria. Plenum, 1982. |
| Weber K, Osborn M: The molecules of the cell matrix. Sci Am 1985;253:110. [PMID: 4071030] |
|






 The cytosolic actin filament. Actin dimers are added to the plus (+) end and removed at the minus (–) end, dynamically lengthening or shortening the filament, as required by the cell. (Redrawn and reproduced, with permission, from Junqueira LC, Carneiro J: Biologia Celular e Molecular, 6th ed. Editora Guanabara, 1997.)
The cytosolic actin filament. Actin dimers are added to the plus (+) end and removed at the minus (–) end, dynamically lengthening or shortening the filament, as required by the cell. (Redrawn and reproduced, with permission, from Junqueira LC, Carneiro J: Biologia Celular e Molecular, 6th ed. Editora Guanabara, 1997.) Molecular organization of a microtubule. In this polarized structure there is an alternation of the two subunits (
Molecular organization of a microtubule. In this polarized structure there is an alternation of the two subunits (









 Three-dimensional representation of a Golgi complex. Through transport vesicles that fuse with the Golgi cis face, the complex receives several types of molecules produced in the rough endoplasmic reticulum (RER). After Golgi processing, these molecules are released from the Golgi trans face in larger vesicles to constitute secretory vesicles, lysosomes, or other cytoplasmic components.
Three-dimensional representation of a Golgi complex. Through transport vesicles that fuse with the Golgi cis face, the complex receives several types of molecules produced in the rough endoplasmic reticulum (RER). After Golgi processing, these molecules are released from the Golgi trans face in larger vesicles to constitute secretory vesicles, lysosomes, or other cytoplasmic components. Electron micrograph of a Golgi complex of a mucous cell. To the right is a cisterna (arrow) of the rough endoplasmic reticulum containing granular material. Close to it are small vesicles containing this material. This is the cis face of the complex. In the center are flattened and stacked cisternae of the Golgi complex. Dilatations can be observed extending from the ends of the cisternae. These dilatations gradually detach themselves from the cisternae and fuse, forming the secretory granules (1, 2, and 3). This is the trans face. Near the plasma membrane of two neighboring cells is endoplasmic reticulum with a smooth section (SER) and a rough section (RER). x30,000. Inset: The Golgi complex as seen in 1-m sections of epididymis cells impregnated with silver. x1200.
Electron micrograph of a Golgi complex of a mucous cell. To the right is a cisterna (arrow) of the rough endoplasmic reticulum containing granular material. Close to it are small vesicles containing this material. This is the cis face of the complex. In the center are flattened and stacked cisternae of the Golgi complex. Dilatations can be observed extending from the ends of the cisternae. These dilatations gradually detach themselves from the cisternae and fuse, forming the secretory granules (1, 2, and 3). This is the trans face. Near the plasma membrane of two neighboring cells is endoplasmic reticulum with a smooth section (SER) and a rough section (RER). x30,000. Inset: The Golgi complex as seen in 1-m sections of epididymis cells impregnated with silver. x1200.
















